Bioinformatics Part 2
Link to part 1
The next problem to solve “Complementing a Strand of DNA” were develop a reverse complement of a DNA string.
Using the reverse_complement function we used before, we can develop the complement using the sample dataset.
DNA_reverse_complement = {'A': 'T', 'T': 'A', 'G': 'C', 'C': 'G'}
def reverse_complement(seq):
return ''.join([DNA_reverse_complement[nuc] for nuc in seq])[::-1]
reverse_complement_string = reverse_complement('AAAACCCGGT')
sample_output = 'ACCGGGTTTT'
if sample_output == reverse_complement_string:
print('MATCH \n')
print('reverse_complement_string: {}'.format(reverse_complement_string))
print('sample_output: {}'.format(sample_output))
else:
print('NO MATCH')I added small if function to check if the result matches the sample dataset.
I got this result:
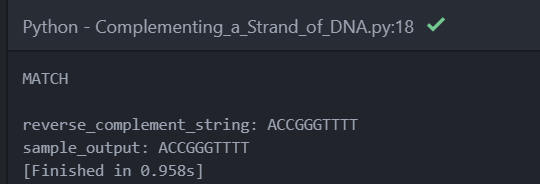
So now we can get the real data:


The code successfully worked:

In the next video, We calculate the GC content. {explain GC content TK}
def gc_content(seq):
return round(seq.count('C') + seq.count('G') / len(seq) * 100)

I checked the GC content on an online tool. I got this result:

Which means my program is undercounting the GC content. I quickly found the issue, which is about the function has missing parenthesis over the seq.count.
Before:
def gc_content(seq):
return round(seq.count('C') + seq.count('G') / len(seq) * 100)After:
def gc_content(seq):
return round((seq.count('C') + seq.count('G')) / len(seq) * 100)Next we make function that calculates the GC content in a subsection.
def gc_content_subsec(seq, k=20):
res = []
for i in range(0, len(seq) - k + 1, k):
subseq = seq[i:i + k]
res.append(gc_content(subseq))
return resNext video to with Rosalind problems again using the function we just made.
The Rosalind problems ask us to work out the GC content. With sting labels in the dataset. This is called the FASTA data format. A widely used format for bioinformatics.

Following the video most of the work is formatting the data so it can be analysed for the GC content.
def read_file(file):
with open(file, 'r') as f:
return [l.strip() for l in f.readlines()]
def gc_content(seq):
return round((seq.count('C') + seq.count('G')) / len(seq) * 100)The first function reads the file. And strips the lines of whitespace. And separates them into lines.
FASTAFile = read_file("rosalind_problems\\test_data\gc_content_ex.txt")
# Dictionary for Labels + Data
FASTADict = {}
# String for holding the current label
FASTALabel = ""
for line in FASTAFile:
if '>' in line:
FASTALabel = line
FASTADict[FASTALabel] = ""
else:
FASTADict[FASTALabel] += lineFor loop adds data into the directory made before. It used an if statement to see the check the open sequence. The “>” symbol is used as the label (key) in the FASTADict. Then others lines are added as values to the key.
RESULTDict = {key: gc_content(value) for (key, value) in FASTADict.items()}
print(RESULTDict)Output:
{'>Rosalind_6404': 54, '>Rosalind_5959': 54, '>Rosalind_0808': 61}Creates a new dictionary containing the GC content of values in the FASTA dict.
MaxGCKey = max(RESULTDict, key=RESULTDict.get) This uses the python max function to find the largest item in the dictionary.
print(f'{MaxGCKey[1:]}\n{RESULTDict[MaxGCKey]}')
The line of code prints the first result of the MaxGCKey. The uses that result in finding the name of the value in the RESULTDict.
Now we can download the dataset form, Rosalind.


Rosalind said to was wrong:

I think the error comes down to the program rounding down/up the result too much. As we can see from the sample output:

The sample out has a lot more decimal points which allow for a finer margining of error. To fix this I simply adjusted the round function to allow 6 decimal points:
def gc_content(seq):
return round((seq.count('C') + seq.count('G')) / len(seq) * 100, 6)
This is the same data from above but allows decimal points to be used.
So I tried the program again with new data:


The program successfully worked:

For the next video, we work on using codons and translation. Codons are a sequence of 3 nucleotides in RNA and DNA strings. Mainly used for protein synthesis. Translations covert the RNA and DNA to proteins.
Copying the codon table turned directory to the structures file:

The translation function:
def translate_seq(seq, init_pos=0):
return [DNA_Codons[seq[pos:pos + 3]] for pos in range(init_pos, len(seq) - 2, 3)][DNA_Codons[seq[pos:pos + 3]] >>> Goes through the DNA_Codons dictionary. The seq variable is the DNA string is inputted. The sequence is then sliced to check 3 nucleotides in the sequence. As the starting position is zero the first nucleotides may look like this:
seq = 'GCATCATTTTCCGACTTGCAGAAGACCTTCTTAGTGAAATTGGGATAACT'
seq[0:3]
'GCA'>>> seq[3:6]
'TCA'for pos in range(init_pos, len(seq) - 2, 3) >>> Starts a for loop with the range of the stating position of the sequence (init_pos variable). Stops the for loop and the end of sequence minus 2. len(seq) - 2. The len(seq) – 2 in the for loop stops the program from reading a sequence that only has 2 nucleotides making it unable to read a codon.
Like this error:

Next function we develop is a function that counts the usage of a codon in a sequence.

def codon_usage(seq, aminoacid):
"""Provides the frequency of each codon encoding a given aminoacid in a DNA sequence"""
tmpList = []
for i in range(0, len(seq) - 2, 3):
if DNA_Codons[seq[i:i + 3]] == aminoacid:
tmpList.append(seq[i:i + 3])
freqDict = dict(Counter(tmpList))
totalWight = sum(freqDict.values())
for seq in freqDict:
freqDict[seq] = round(freqDict[seq] / totalWight, 2)
return freqDicttmpList = [] >>> creates temporary list to codonsfor i in range(0, len(seq) - 2, 3): >>> Makes loop that starts at 0 and then with the length of the sequence -2 with the increment of 3.if DNA_Codons[seq[i:i + 3]] == aminoacid:
tmpList.append(seq[i:i + 3])If statement checks if codon matches with amino acid selected in the Function. If the codon matches the amino acid then the codon is then added to the temporary list.
freqDict = dict(Counter(tmpList)) >>> creates a dictionary with amount of times the codon occurred in the list.
totalWight = sum(freqDict.values()) >>> Adds up all the number together so it later be used to calculate the percent of amino acids
for seq in freqDict:
freqDict[seq] = round(freqDict[seq] / totalWight, 2)A small for loop that calculates the percentage of the codon. By dividing the frequency of codon with the total number codons with totalWight. The rounding by decimal points.